

General Research Interests
My research program aims to understand how species adapt to environmental stressors such as climate change, pathogens, and invasive species, and how they respond to ongoing pressures such as habitat loss. I integrate field experiments, population and comparative genomics, and computational modeling to uncover the molecular mechanisms driving adaptation and resilience, as well as the evolutionary processes that drive population and species differences. Focal species of my research program range from forest trees and their associated pests and pathogens, to Arctic willow and salmonid species, to several rare species of high conservation priority such as the red-cockaded woodpecker, longleaf pine, and whitebark pine. A central goal of my research is to translate genomic insights into practical tools for biodiversity conservation and ecosystem management. By bridging molecular biology, ecology, and data science, my work addresses both basic and applied outcomes for sustaining biodiversity in a changing world.
Research Projects
Training future scientists through conservation genomics: longleaf pine and red-cockaded woodpecker genome projects
In collaboration with the University of Florida, the US Fish & Wildlife Service, and Archbold Biological Station, I am directing the assembly of the first reference genomes for two ecologically critical and imperiled species: the endangered longleaf pine (Pinus palustris) and the threatened red-cockaded woodpecker (Dryobates borealis). These species co-occur in fire-adapted Southeastern U.S. ecosystems, where the red-cockaded woodpecker is dependent upon mature longleaf pine stands. Supported by funding awarded through ORG.one, this project has been integrated into the Biology and Conservation Genomics undergraduate training program at UConn - a year-long program that trains several undergraduate students in bioinformatics, genome assembly, and conservation biology. By actively engaging with conservation practitioners, students gain hands-on learning while contributing critical genomic resources to support species recovery and ecosystem management. At the end of the program the students will publish a peer-reviewed article describing the assembly and insights gained through comparative genomics.The Evolving Meta-ecosystems Biology Integration Institute
As part of the NSF-funded Evolving Meta-Ecosystems (EVOME) Biology Integration Institute, my research explores how rapid climate change shapes evolutionary processes and species interactions across Arctic river and tundra ecosystems—some of the most rapidly warming systems on Earth. I recently wrote about the perspective piece for the American Journal of Botany's On the Nature of Things series describing the EVOME framework to connect genes to ecosystems. My work for the Institute focusses on two keystone species: Arctic grayling (Thymallus arcticus) and feltleaf willow (Salix alaxensis), both of which are locally adapted and central to Arctic community dynamics. Using long-read sequencing, I am assembling the first reference genomes for these species and several related willows to investigate conserved regions, gene family evolution, and structural variation. To better capture species-level diversity, I am constructing reference-aided pangenome graphs that integrate landscape genomic sampling and common garden phenotypes. Ultimately, this work contributes to trait-based predictions within ecological models, offering insight into how evolutionary processes shape the structure and function of complex ecosystems.I am also leading data management across the Institute and work closely with the BLM and Toolik staff. This involves the design of data collection surveys using user-friendly, barcode-enabled field and lab apps such as Kobo Collect, TreeSnap, and AppSheet, the ingestion and integration of data within and outside the Institute, data reporting to NSF, and ultimately final data archiving. However, institutes, field stations, and research centers often face challenges due to disconnected data sources, inconsistent metadata, and limited tools for synthesis. In my most recent manuscript, we describe CartograPlant – a web-based platform for integrating genomic, phenotypic, and environmental data. By developing scalable informatics pipelines that enforce FAIR principles and real-time analysis through high-performance computing, we created a system that now hosts over 25 million georeferenced plant records across 846 species, cultivars, and hybrid complexes, as well as almost 1000 environmental layers including datasets from BIEN, NEON, LTER, and FIA. We have begun incoporating data from EVOME, which had its first full field season in 2025. Together, these efforts strengthen our capacity to integrate complex datasets and uncover how evolutionary processes contribute to ecosystem resilience in the Arctic and beyond.
The accuracy of predicting maladaptation to new environments with genomic data
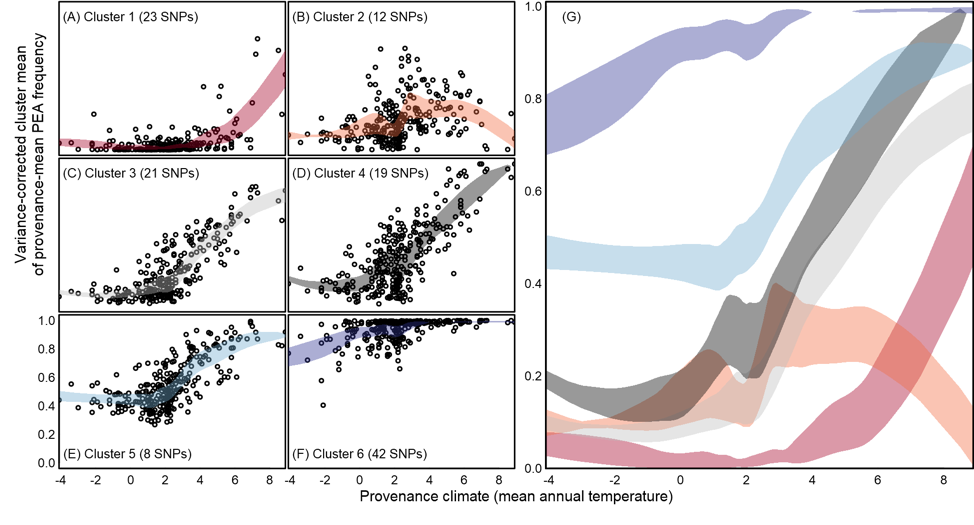
Methods using genomic information to forecast population maladaptation to climate change (i.e., genetic offset methods) are becoming increasingly common, yet there has been little exploration into the factors that could potentially affect performance of these methods, or mislead inference with regard to model accuracy during validation. These knowledge gaps pose serious hurdles toward the incorporation of such methods into management and policy. In Dr. Lotterhos' lab at Northeastern, I used extensive sets of simulations to understand the circumstances in which these methods will be most accurate. We found several factors related to the evolutionary history of targeted populations, as well as the experimental design used to train models, that affect model accuracy. Our work was featured in Molecular Ecology Resources on the issue's cover, and is part of a special issue in the American Naturalist.The CoAdapTree Project
My first postdoc was at the University of British Columbia working on the CoAdapTree project with Sally Aitken and Sam Yeaman. This project examines how biotic (eg pathogens) and abiotic (eg climate) factors influence genomic variation across four conifer species. My primary responsibilities focussed on developing genetic resources to describe the adaptive genetics of fungal- (e.g., swiss needle cast and rhabdocline and dothistroma needle blight), cold-, and drought-hardiness in 70 Douglas-fir and 87 western Larch populations, as well as climate adaptation in these two species, lodgepole pine, and jack pine (see map to left). To handle all of this data, and to do so with reproducibility in mind, I've made my bioinformatics pipeline publically available. In fact, I've made all of the code from each lead-authored project available on GitHub, including our case-control genotype-phenotype method. See Data Availability statements within each manuscript.The genetic architecture of repeated local adaptation to climate in distantly related plants PDF
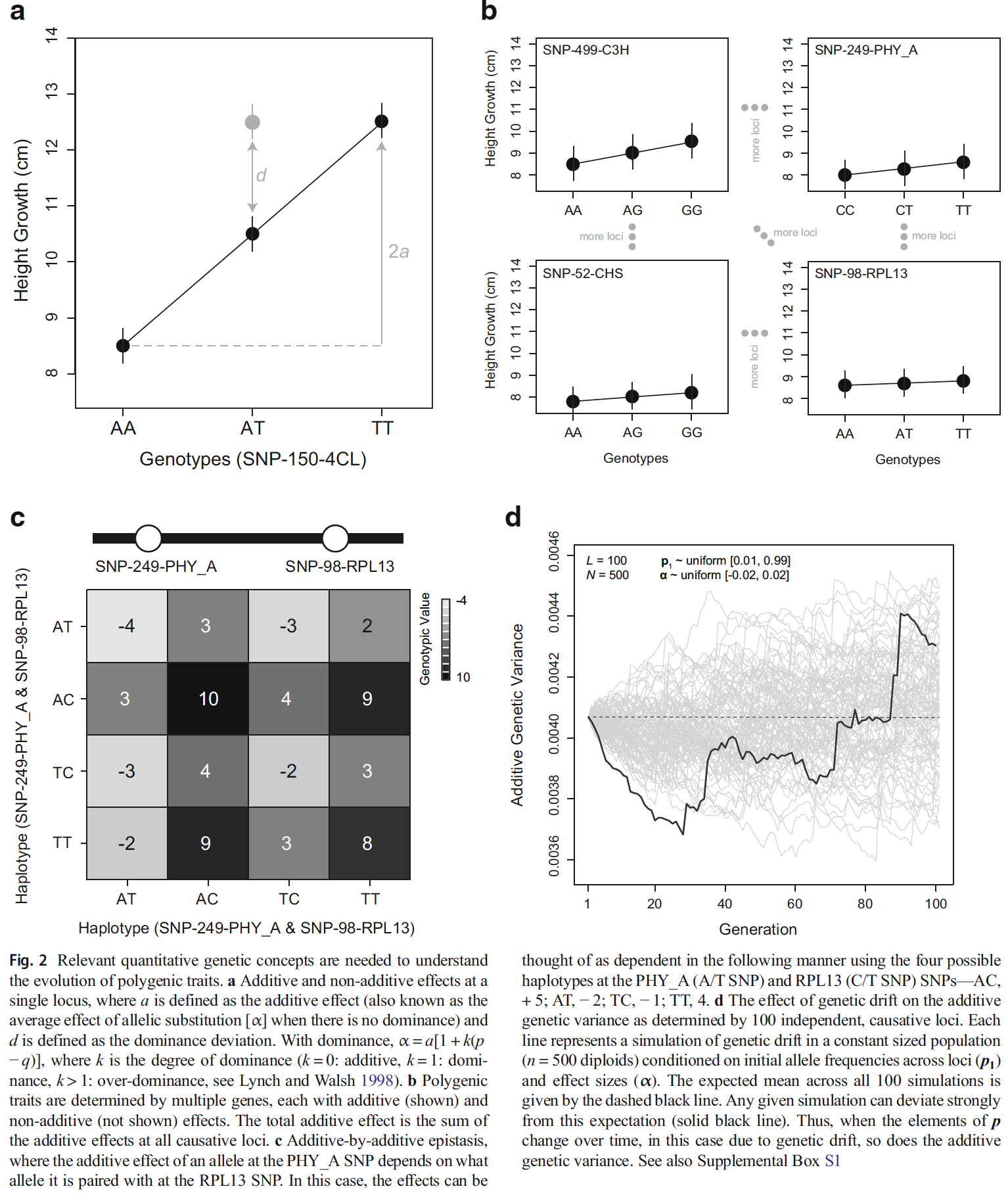
How repeatable is evolution? This question, popularized by Stephen Jay Gould’s ‘replaying the tape of life’ thought experiment, has been the subject of decades of empirical research. Reanalyzing genomic data from thousands of individuals across 25 plant species, I contributed to a study that identified 108 gene families repeatedly linked to climate adaptation. These genes will be crucial for monitoring in breeding and natural populations to assess the how well populations are maintaining climate adaptation. This study also highlights the advantage of phylogenetic analyses to understand adaptation.How useful is genomic data for predicting maladaptation to future climate? PDF
The pace of climate change threatens to decouple adaptive variation and environmental optima of many natural populations. Characterising the vulnerability of populations across a species range can be particularly useful to direct management priorities. Over the past decade, using genomic data to predict maladaptation of natural populations to future climate (i.e., genomic offset methods) have become increasingly widespread but with few instances where model predictions are validated with ground truth data. We use range-wide data across three conifer taxa to assess such methods. We validate these methods against two-year and 52-year growth and mortality measured in independent transplant experiments. Overall, we find that two common methods often better predict transplant performance than climatic or geographic distances. We also find that these models are surprisingly not improved using GEA candidates. Even with promising validation results, variation in model projections to future climates can make it difficult to identify the most maladapted populations using either method.
Evaluating genomic data for management of local adaptation in a changing climate PDF
Understanding how managed populations are adapted to their local environments will assist in both breeding and reforestation efforts by identifying environmental sources driving trait differentiation and pathogen susceptibility. To feasibly (weighing time, effort, and cost) disect these aspects of plant biology, there are a few common approaches that can be considered, but do they hold the same information content with regard to the environmental drivers of differentiation or the spatial scale of adaptation? To address this, we compared climate data, genomic data, and a long term provinance trial in lodgepole pine in this regard. We discuss relative strengths and weaknesses when considering the appropriate data for managing populations with broad geographic ranges. (see figure to the left showing range-wide clines in genomic regions underlying adaptation)
Evolutionary genomics of the invasive and defoliating gypsy moth PDF
Biological invasions from non-native species can have have large economic consequences and can fundamentally alter the ecology of the invaded system. As one such example, the European gypsy moth was introduced to the eastern United States around the late 19th century and has since spread north through Maine, west through Wisconsin, and south through the Carolinas. Consequently, the gypsy moth has become one of the most destructive hardwood pests in North America. To understand how this species has been so successful in a novel environment we sampled populations from the range edges by examining genomic variation underlying trait differentiation for three developmental traits. We described how the invasion was facilitated through adaptive variation found throughout the genome and that genomic patterns (ie linkage disequilibrium) among adaptive genetic componants were driving differences in the three traits among populations. (see map to the left showing the chronological spread of the gypsy moth across New England)
Effects of Forest Management on Fine-scale Gene Flow in a Fire-suppressed population of Pinus lambertiana Dougl. PDF

Effect of fire supression on forest crowding in the upper Yosemite Valley from Columbia Point (source unknown)
Climate change is exacerbating the effects of fire suppression in the west, and prescribed fire and forest thinning are two ways of mitigating these effects. Sugar pine is an important species in these forests, and was historically dominant before fire suppression. We looked at how these treatments affect mating patterns in this species and discuss how this may affect long-term forest dynamics. (see 3D stem map to the left; more about Teakettle Experimental Forest here).
Local adaptation of the endangered whitebark pine across fine spatial scales of the Lake Tahoe Basin - PDF
Whitebark pine (Pinus albicaulis) is a foundational species in many high elevation forests and plays an important role in ecosystem function and services. Despite its influential role in these systems, whitebark pine is listed as a threatened species by IUCN. Therefore, understanding the spatial scale of adaptation in this species is vital for conservation efforts. Even so, much of our knowledge regarding adaptation in trees comes from broad geographic ranges, often beyond the scale at which most forest management is applied. Using a common garden we found that whitebark pine is adapted across the fine spatial scales of the Lake Tahoe Basin despite highly shared genetic variation across populations. We further showed that the adaptive signal driven by water availability is apparent across common garden and genomicw data, likely due to the rain shadow on the eastern Siera Nevada. (to the left see map of sampled populations from the Lake Tahoe Basin, and how quickly the rainshadow reduces annual preciptation by 50% in just under 15 miles)
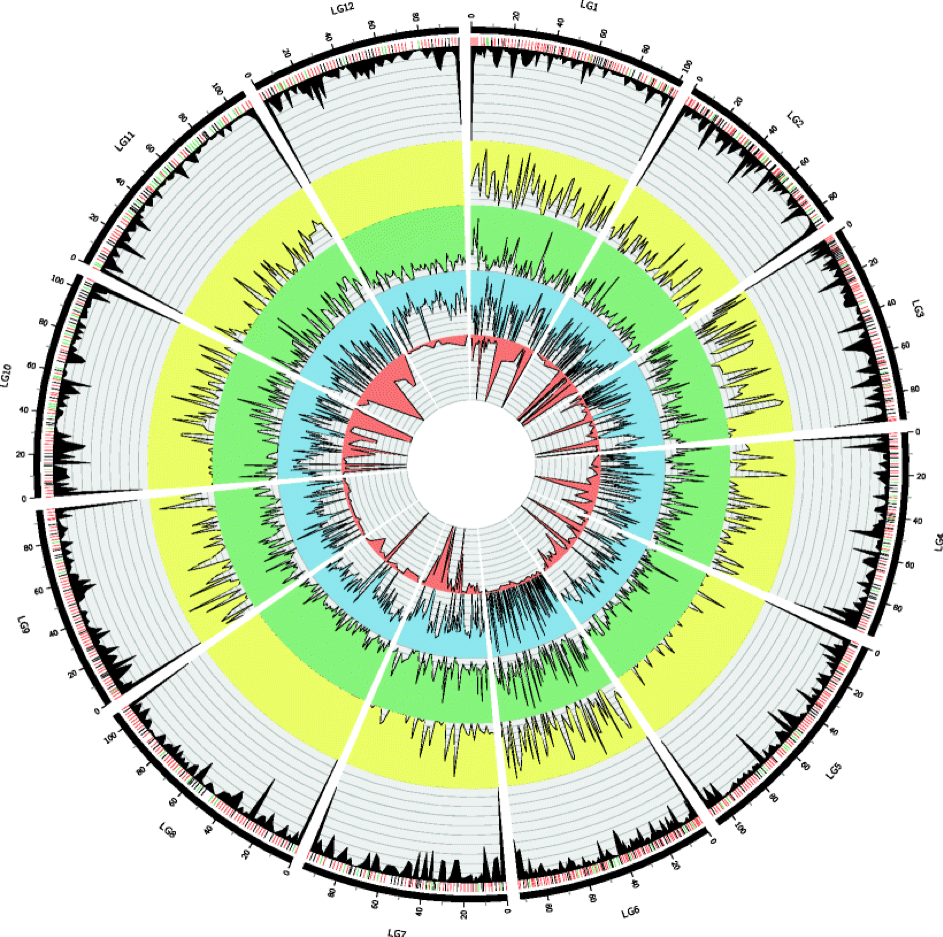
Linkage maps and the genomic architecture of local adaptation
For non-model organisms, genomic resources needed to fully describe the genetic and genomic architectures of complex traits can face many hurdles. Because of their large genome size, conifers face many challenges in this regard, particularly with respect to the production of physical sequence maps with ordered genetic markers. Such resources can be used to test fundamental theory regarding the expected genetic architecture underlying adaptive traits under various demographic and evolutionary scenarios. To produce such resources, linkage maps are often used to both order and elongate blocks of sequence data. Across taxa, such maps were predominately made from well understood familial relationships or mating designs, but maps of single trees began increasing in numbers in the 1990s. In conifers, maternally derived haploid tissue (i.e., a single copy of the genome from megagametophye tissue) can be used to order markers within individual trees within a fairly straightforward framework. To begin to test fundamental theories of local adaptation in a non-model conifer, Pinus balfouriana, we constructed a dense linkage map using individuals collected from populations that historically exhibited gene flow but have subsequently become isolated from one another since the last glacial maximum. PDF
After completing this linkage map, we used niche modeling and indirect measures of WUE, the ratio of carbon isotopes fixed during photosynthesis (δ13C), as well as nutrient utilization, the ratio of nitrogen isotopes fixed during leaf-level resource utilization (μgN), to explore the importance of water availability as a determinant of geographical range of foxtail pine. Additionally, we explored the degree to which variation in δ13C and μgN is genetically based, differentiated among populations, and underlain by a genetic architecture with large effect loci. PDF



{kind=link}